
Isotopic Radiolabeling of the Antiretroviral Drug [18F]Dolutegravir for Pharmacokinetic PET Imaging

 , , , ,
, , , ,
Abstract
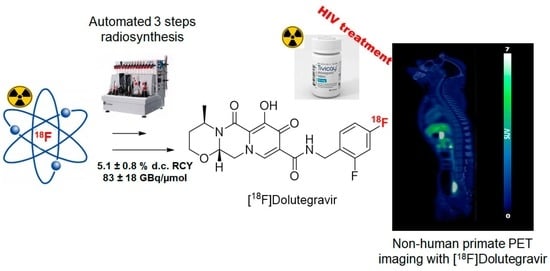
:
1. Introduction
2. Results and Discussion
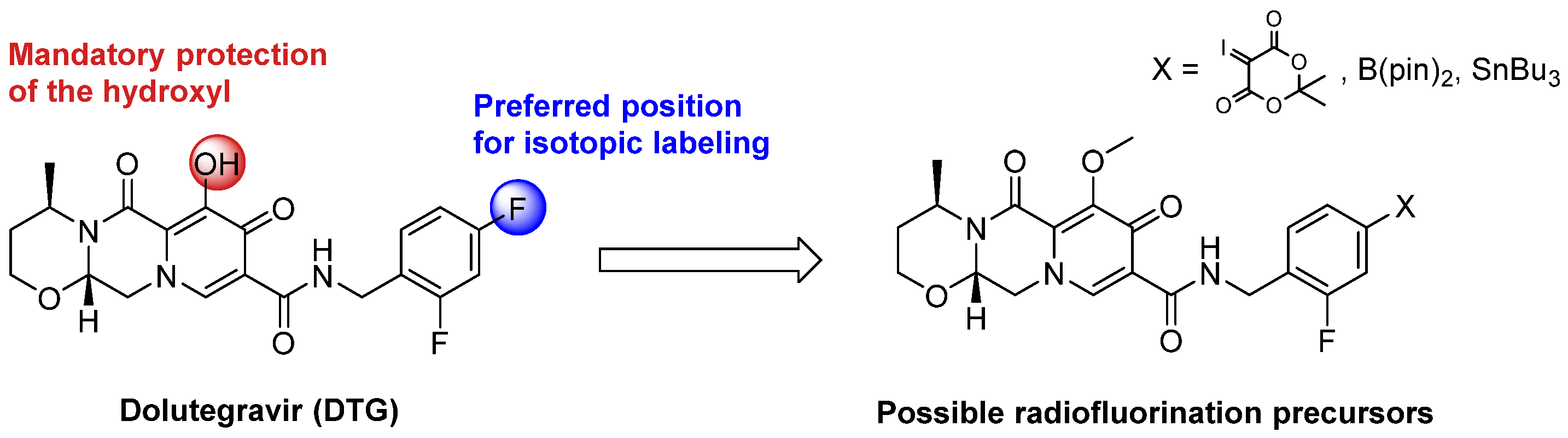
2.1. Late-Stage Radiolabeling Approach
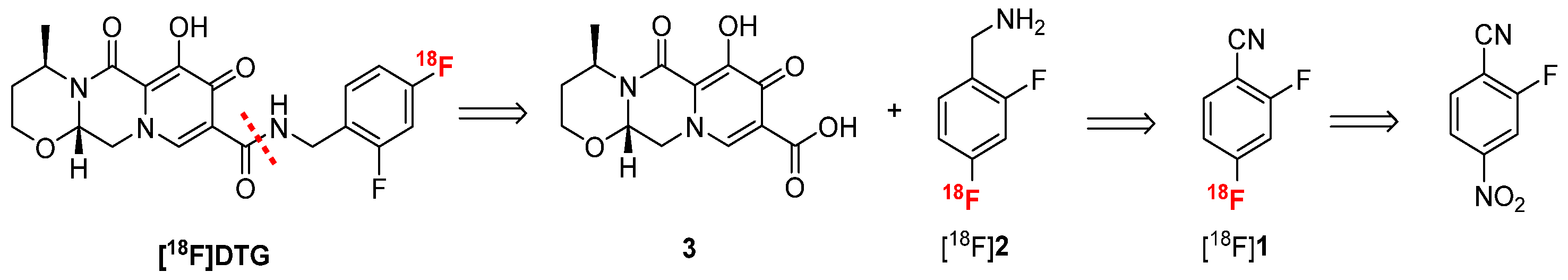
2.2. Three-Step Radiolabeling Approach
2.2.1. Radiofluorination Optimization
2.2.2. Nitrile Reduction Optimization
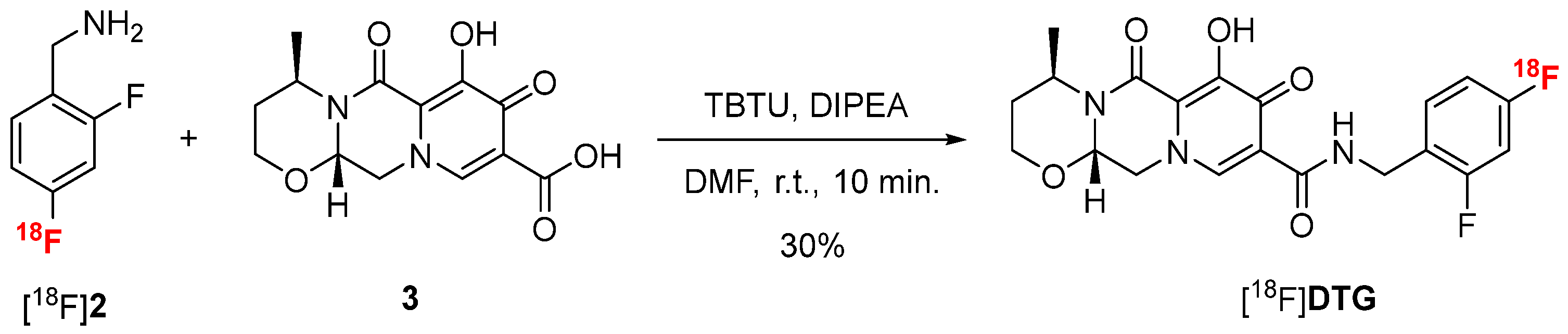
2.2.3. Peptide Coupling Optimization
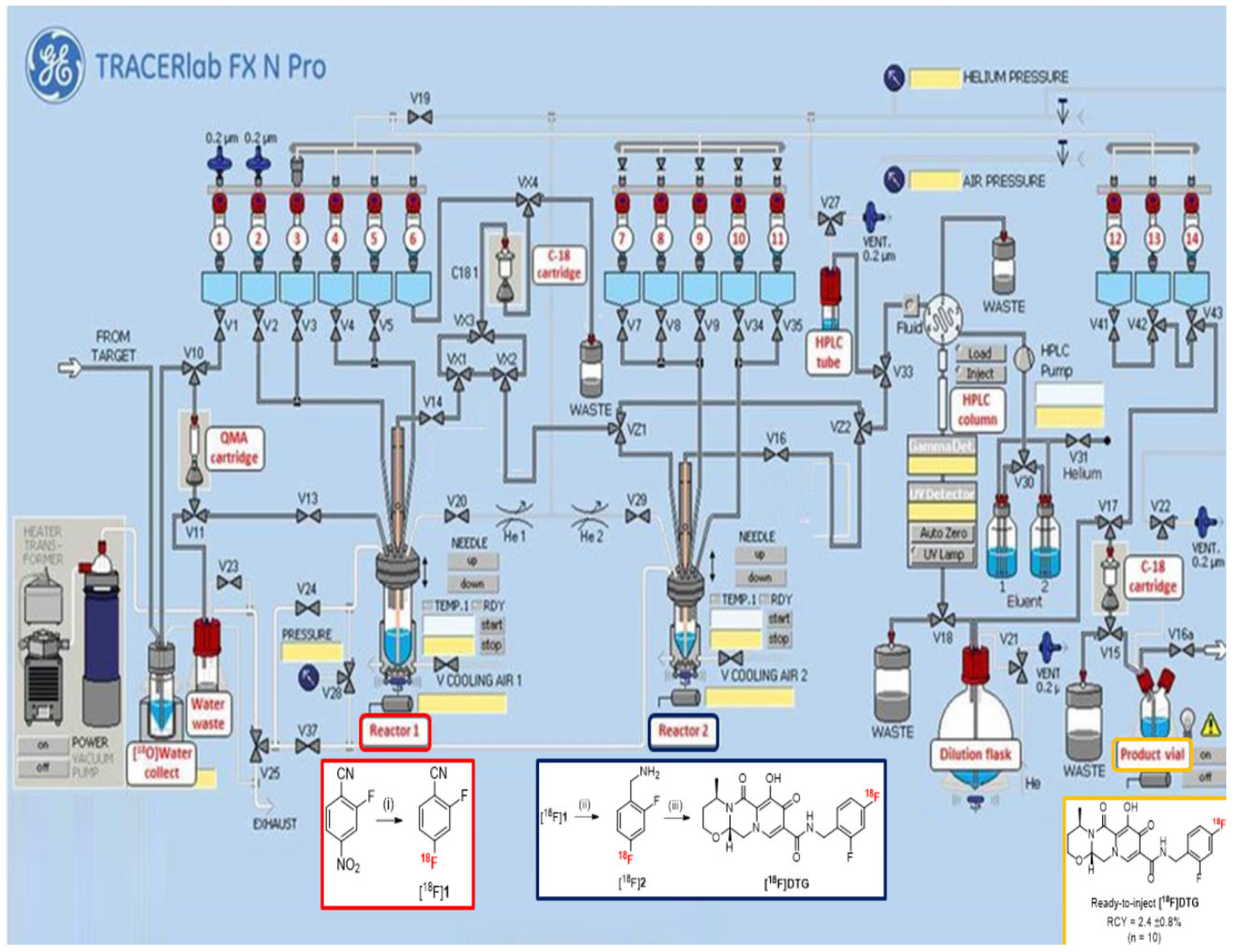
2.3. Automated Radiosynthesis of [18F]DTG
2.4. Quality Control
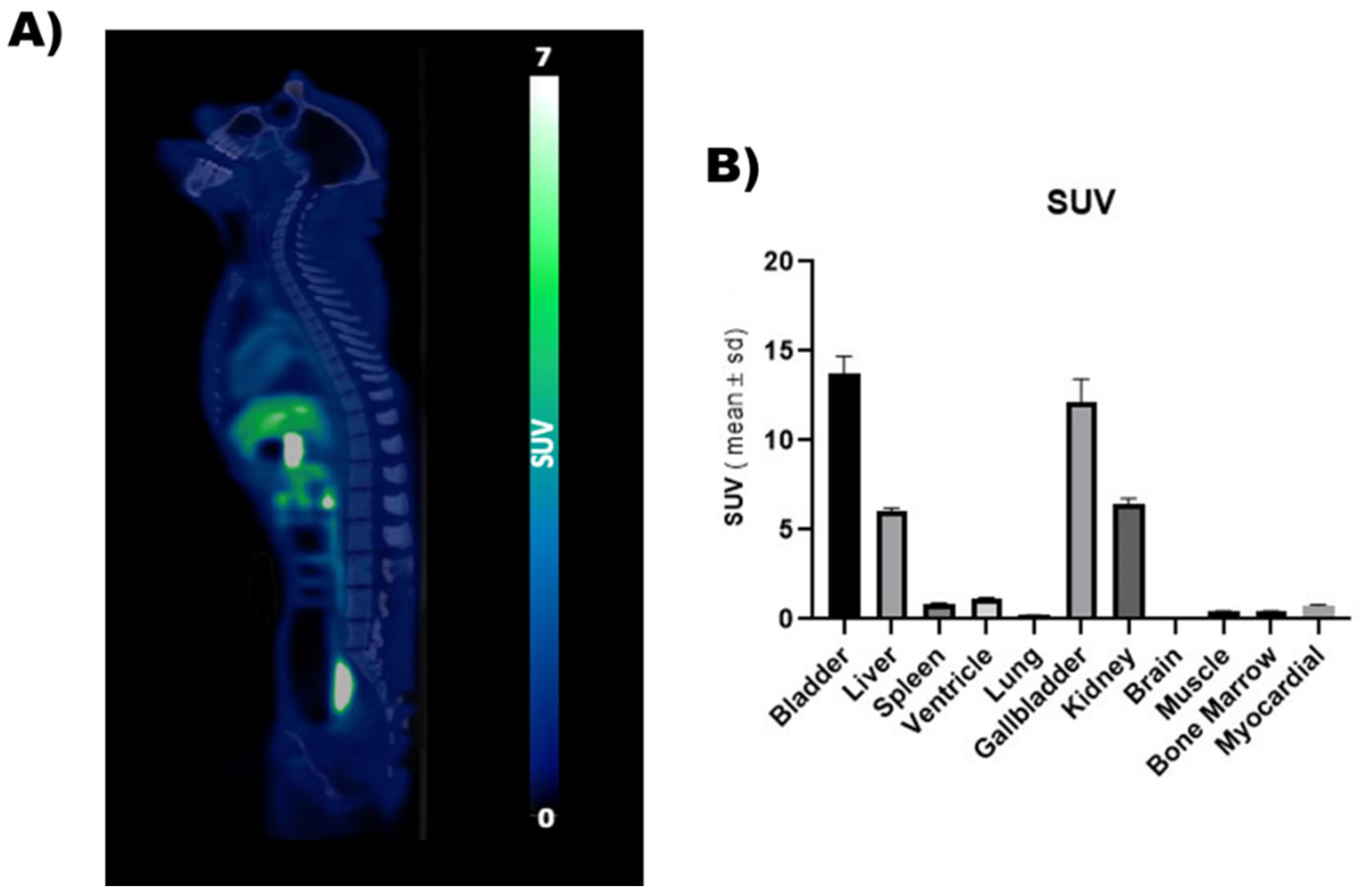
2.5. PET Imaging
3. Materials and Methods
3.1. Chemistry
3.2. Radiochemistry
3.3. PET Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-Treatment HIV-1 Controllers with a Long-Term Virological Remission after the Interruption of Early Initiated Antiretroviral Therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Cirión, A.; Pancino, G. HIV controllers: A genetically determined or inducible phenotype? Immunol. Rev. 2013, 254, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Passaes, C.P.; Sáez-Cirión, A. HIV cure research: Advances and prospects. Virology 2014, 454, 340–352. [Google Scholar] [CrossRef] [PubMed]
- The Antiretroviral Therapy Cohort Collaboration. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: A collaborative analysis of 14 cohort studies. Lancet 2008, 372, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Cory, T.J.; Schacker, T.W.; Stevenson, M.; Fletcher, C.V. Overcoming pharmacologic sanctuaries. Curr. Opin. HIV AIDS 2013, 8, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, H.-H.; Lichterfeld, M. Recent progress in understanding HIV reservoirs. Curr. Opin. HIV AIDS 2018, 13, 137–142. [Google Scholar] [CrossRef]
- Di Mascio, M.; Srinivasula, S.; Bhattacharjee, A.; Cheng, L.; Martiniova, L.; Herscovitch, P.; Lertora, J.; Kiesewetter, D. Antiretroviral tissue kinetics: In vivo imaging using positron emission tomography. Antimicrob. Agents Chemother. 2009, 53, 4086–4095. [Google Scholar] [CrossRef] [Green Version]
- Di Mascio, M.; Paik, C.H.; Carrasquillo, J.A.; Maeng, J.-S.; Jang, B.-S.; Shin, I.S.; Srinivasula, S.; Byrum, R.; Neria, A.; Kopp, W.; et al. Antiretroviral Tissue Kinetics: In Vivo Imaging Using Positron Emission Tomography. Blood 2009, 114, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, P.J.; Rogers, K.A.; Zurla, C.; Blanchard, E.L.; Gumber, S.; Strait, K.; Connor-Stroud, F.; Schuster, D.M.; Amancha, P.K.; Hong, J.J.; et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiretroviral therapy-treated macaques. Nat. Methods 2015, 12, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Henrich, T.J.; Hsue, P.Y.; VanBrocklin, H. Seeing Is Believing: Nuclear Imaging of HIV Persistence. Immunology 2019, 10, 2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronald, J.A.; Kim, B.-S.; Gowrishankar, G.; Namavari, M.; Alam, I.S.; D’Souza, A.; Nishikii, H.; Chuang, H.-Y.; Ilovich, O.; Lin, C.-F.; et al. A PET Imaging Strategy to Visualize Activated T Cells in Acute Graft-versus-Host Disease Elicited by Allogenic Hematopoietic Cell Transplant. Cancer Res. 2017, 77, 2893–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namavari, M.; Chang, Y.-F.; Kusler, B.; Yaghoubi, S.; Mitchell, B.S.; Gambhir, S.S. Synthesis of 2′-Deoxy-2′-[18F]Fluoro-9-β-D-Arabinofuranosylguanine: A Novel Agent for Imaging T-Cell Activation with PET. Mol. Imaging Biol. 2011, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Rosenkrans, Z.T.; Liu, J.; Huang, G.; Luo, Q.-Y.; Cai, W. ImmunoPET: Concept, Design, and Applications. Chem. Rev. 2020, 120, 3787–3851. [Google Scholar] [CrossRef]
- McMahon, J.H.; Zerbato, J.M.; Lau, J.S.Y.; Lange, J.L.; Roche, M.; Tumpach, C.; Dantanarayana, A.; Rhodes, A.; Chang, J.; Rasmussen, T.A.; et al. A clinical trial of non-invasive imaging with an anti-HIV antibody labelled with copper-64 in people living with HIV and uninfected controls. EBioMedicine 2021, 65, 103252. [Google Scholar] [CrossRef]
- Vera, D.B.; Schulte, B.; Henrich, T.; Flavell, R.; Seo, Y.; Abdelhafez, Y.; Badawi, R.; Cherry, S.; VanBrocklin, H. First-in-human total-body PET imaging of HIV with 89Zr-VRC01 on the EXPLORER. J. Nucl. Med. 2020, 61, 545. [Google Scholar]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef]
- Sax, P.E.; Pozniak, A.; Montes, M.L.; Koenig, E.; DeJesus, E.; Stellbrink, H.-J.; Antinori, A.; Workowski, K.; Slim, J.; Reynes, J.; et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380–1490): A randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet 2017, 390, 2073–2082. [Google Scholar]
- Cahn, P.; Pozniak, A.L.; Mingrone, H.; Shuldyakov, A.; Brites, C.; Andrade-Villanueva, J.F.; Richmond, G.; Buendia, C.B.; Fourie, J.; Ramgopal, M.; et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: Week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 2013, 382, 700–708. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/news/item/22-07-2019-who-recommends-dolutegravir-as-preferred-hiv-treatment-option-in-all-population (accessed on 10 January 2022).
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef] [Green Version]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem. Eur. J. 2015, 21, 5972–5979. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Cheng, R.; Chen, P.; Liu, G.; Liang, S.H. Novel Path to Aryl(isoquinoline)iodonium(III) Salts and Synthesis of Radiofluorinated Isoquinolines. Angew. Chem. Int. Ed. 2016, 55, 11882–11886. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, B.H.; Stephenson, N.A.; Vasdev, N.; Liang, S.H. Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun. 2014, 5, 4365. [Google Scholar] [CrossRef] [Green Version]
- Rotstein, B.H.; Wang, L.; Liu, R.Y.; Patteson, J.; Kwan, E.E.; Vasdev, N.; Liang, S.H. Mechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(III) ylides. Chem. Sci. 2016, 7, 4407–4417. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, J.E.; Grønnevik, G.; Riss, P.J. Organocatalyst-assisted Ar–18F bond formation: A universal procedure for direct aromatic radiofluorination. Chem. Commun. 2017, 53, 12906–12909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossine, A.V.; Brooks, A.F.; Makaravage, K.J.; Miller, J.M.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org. Lett. 2015, 17, 5780–5783. [Google Scholar] [CrossRef] [PubMed]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem. Int. Ed. 2014, 53, 7751–7755. [Google Scholar] [CrossRef]
- Antuganov, D.; Zykov, M.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Krasikova, R. Effect of Pyridine Addition on the Efficiency of Copper-Mediated Radiofluorination of Aryl Pinacol Boronates. ChemistrySelect 2017, 2, 7909–7912. [Google Scholar] [CrossRef]
- Taylor, N.J.; Emer, E.; Preshlock, S.; Schedler, M.; Tredwell, M.; Verhoog, S.; Mercier, J.; Genicot, C.; Gouverneur, V. Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc. 2017, 139, 8267–8276. [Google Scholar] [CrossRef]
- Zhang, X.; Basuli, F.; Swenson, R.E. Positron Emission Tomography Imaging with 2-[18F]F-p-Aminobenzoic Acid Detects Staphylococcus aureus Infections and Monitors Drug Response. J. Label. Compd. Radiopharm. 2019, 62, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef] [PubMed]
- Lahdenpohja, S.; Keller, T.; Rajander, J.; Kirjavainen, A.K. Radiosynthesis of the norepinephrine transporter tracer [18F]NS12137 via copper-mediated 18F-labelling. J. Label. Compd. Radiopharm. 2019, 62, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Goodman, S.N.; Mans, D.; Kowalski, M. Process for Preparing Carbamoylpyridone Derivatives and Intermediates. EP3260457A1, 27 December 2017. [Google Scholar]
- Li, W.; Thompson, W.; Fisher, T.; Wai, J.S.; Hazuda, D.; Burns, H.D.; Hamill, T.G. Radiosynthesis of the HIV integrase inhibitor [18F]MK-0518 (Isentress). J. Label. Compd. Radiopharm. 2010, 53, 517–520. [Google Scholar] [CrossRef]
- Blecha, J.; Neumann, K.; VanBrocklin, H. Automated synthesis of [18F]Raltegravir through [18F]fluorobenzylamine. J. Nucl. Med. 2018, 59, 669. [Google Scholar]
- Dietz, J.-P.; Lucas, T.; Groß, J.; Seitel, S.; Brauer, J.; Ferenc, D.; Gupton, B.F.; Opatz, T. Six-Step Gram-Scale Synthesis of the Human Immunodeficiency Virus Integrase Inhibitor Dolutegravir Sodium. Org. Process Res. Dev. 2021, 25, 1898–1910. [Google Scholar] [CrossRef]
- van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [Green Version]
- Koslowsky, I.; Mercer, J.; Wuest, F. Synthesis and application of 4-[18F]fluorobenzylamine: A versatile building block for the preparation of PET radiotracers. Org. Biomol. Chem. 2010, 8, 4730–4735. [Google Scholar] [CrossRef]
- Clot, E.; Eisenstein, O.; Jasim, N.; Macgregor, S.A.; McGrady, J.E.; Perutz, R.N. C−F and C−H Bond Activation of Fluorobenzenes and Fluoropyridines at Transition Metal Centers: How Fluorine Tips the Scales. Acc. Chem. Res. 2011, 44, 333–348. [Google Scholar] [CrossRef]
- Lv, H.; Cai, Y.-B.; Zhang, J.-L. Copper-Catalyzed Hydrodefluorination of Fluoroarenes by Copper Hydride Intermediates. Angew. Chem. Int. Ed. 2013, 52, 3203–3207. [Google Scholar] [CrossRef]
- Burkhardt, E.R.; Matos, K. Boron Reagents in Process Chemistry: Excellent Tools for Selective Reductions. Chem. Rev. 2006, 106, 2617–2650. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, L.; Máthé, T. Selective heterogeneous catalytic hydrogenation of nitriles to primary amines in liquid phase: Part I. Hydrogenation of benzonitrile over palladium. Appl. Catal. Gen. 2005, 296, 209–215. [Google Scholar] [CrossRef]
- Gair, J.J.; Grey, R.L.; Giroux, S.; Brodney, M.A. Palladium Catalyzed Hydrodefluorination of Fluoro-(hetero)arenes. Org. Lett. 2019, 21, 2482–2487. [Google Scholar] [CrossRef] [PubMed]
- Werkmeister, S.; Bornschein, C.; Junge, K.; Beller, M. Selective Ruthenium-Catalyzed Transfer Hydrogenations of Nitriles to Amines with 2-Butanol. Chem. Eur. J. 2013, 19, 4437–4440. [Google Scholar] [CrossRef] [PubMed]
- Caddick, S.; Judd, D.B.; Lewis, A.K.D.K.; Reich, M.T.; Williams, M.R.V. A generic approach for the catalytic reduction of nitriles. Tetrahedron 2003, 59, 5417–5423. [Google Scholar] [CrossRef]
- Way, J.; Wuest, F. Fully automated synthesis of 4-[18F]fluorobenzylamine based on borohydride/NiCl2 reduction. Nucl. Med. Biol. 2013, 40, 430–436. [Google Scholar] [CrossRef]
- Tietz, O.; Sharma, S.K.; Kaur, J.; Way, J.; Marshall, A.; Wuest, M.; Wuest, F. Synthesis of three 18F-labelled cyclooxygenase-2 (COX-2) inhibitors based on a pyrimidine scaffold. Org. Biomol. Chem. 2013, 11, 8052–8064. [Google Scholar] [CrossRef]
- Budidet, S.R.; Dussa, N.; Kaki, G.R.; Yatcherla, S.R.; Sanapureddy, J.M.R.; Danda, S.R.; Katuroju, S.; Meenakshisunderam, S. An Improved Process for the Preparation of Dolutegravir. International Patent Application No. WO2014128545A2, 28 August 2014. [Google Scholar]
- Kalantzopoulos, G.N.; Guzik, M.N.; Deledda, S.; Heyn, R.H.; Muller, J.; Hauback, B.C. Destabilization effect of transition metal fluorides on sodium borohydride. Phys. Chem. Chem. Phys. 2014, 16, 20483–20491. [Google Scholar] [CrossRef]
- General Monograph 0125—Radiopharmaceutical Preparations in European Pharmacopoeia; Council of Europe: Strasbourg, France, 2016.
- Castellino, S.; Moss, L.; Wagner, D.; Borland, J.; Song, I.; Chen, S.; Lou, Y.; Min, S.S.; Goljer, I.; Culp, A.; et al. Metabolism, Excretion, and Mass Balance of the HIV-1 Integrase Inhibitor Dolutegravir in Humans. Antimicrob. Agents Chemother. 2013, 57, 3536–3546. [Google Scholar] [CrossRef] [Green Version]
- Labarthe, L.; Gelé, T.; Gouget, H.; Benzemrane, M.-S.; Le Calvez, P.; Legrand, N.; Lambotte, O.; Le Grand, R.; Bourgeois, C.; Barrail-Tran, A. Pharmacokinetics and tissue distribution of tenofovir, emtricitabine and dolutegravir in mice. J. Antimicrob. Chemother. 2022, 77, 1094–1101. [Google Scholar] [CrossRef]
- Tournier, N.; Saba, W.; Cisternino, S.; Peyronneau, M.-A.; Damont, A.; Goutal, S.; Dubois, A.; Dollé, F.; Scherrmann, J.-M.; Valette, H.; et al. Effects of Selected OATP and/or ABC Transporter Inhibitors on the Brain and Whole-Body Distribution of Glyburide. AAPS J. 2013, 15, 1082–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
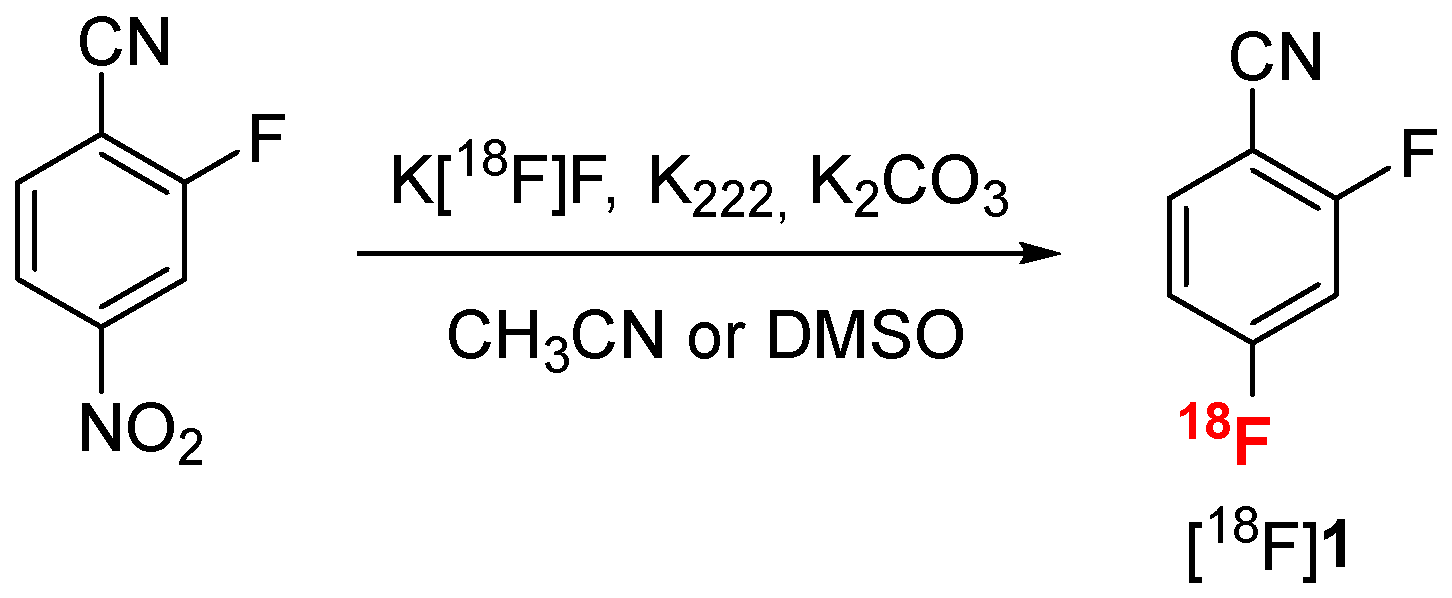{kind=link}
{kind=link}
| Entry 1 | Solvent | Reaction Time | Conversion in [18F]1 2 |
|---|---|---|---|
| 1 | CH3CN | 5 min | 96% |
| 2 | CH3CN | 10 min | 97% |
| 3 | CH3CN | 15 min | 97% |
| 4 | DMSO | 5 min | 90% |
 | ||||
|---|---|---|---|---|
| Entry 1 | Reducing Agents | Reaction Conditions | [18F]2 Conversion 2 | Side Products Conversion 2 |
| 1 | LAH (100 µmol) | THF, r.t., 5min | 15% | 85% |
| 2 | LAH (100 µmol) | THF, 50 °C, 5min | 50% | 50% |
| 3 | BH3.THF (200 µmol) | THF, r.t., 15 min | 5% | 91% |
| 4 | BH3.THF (200 µmol) | THF, 65 °C, 5 min | 5% | 91% |
| 5 | H2 (1 atm) | Pd/C (150 µg), MeOH, r.t., 5 min | 0% | 30% |
| 6 | [RuCl2(p-cymene)2] (1.5 µmol) | DPPP, NaOH 3M iPrOH, 100 °C, 15 min | 10% | - |
| 7 | NaBH4 (400 µmol), NiCl2 6H2O (4 µmol) | MeOH, r.t., 5 min | 42% | 22% |
| 8 | BER (500 mg), NiCl2 6H2O (20 mg) | MeOH | <5% | - |
| Entry 1 | NaBH4 (µmol) | NiCl2·6H2O (µmol) | H2O (µL) | T (°C) | Time (min) | [18F]2 Conversion 2 | Side Products Conversion 3 |
|---|---|---|---|---|---|---|---|
| 1 | 400 | 4 | - | r.t. | 5 | 42% | 22% |
| 2 | 40 | 4 | - | r.t. | 5 | 20% | 80% |
| 3 | 40 | 4 | - | r.t. | 10 | 23% | 77% |
| 4 | 40 | 4 | - | 50 °C | 5 | 26% | 74% |
| 5 | 40 | 4 | 100 | r.t. | 5 | 45% | 55% |
| 6 | 40 | 20 | 100 | r.t. | 5 | 47% | 53% |
| 7 | 40 | 40 | 100 | r.t. | 5 | 41% | 59% |
| 8 | 40 | 100 | 100 | r.t. | 5 | 72% | 28% |
| 9 | 40 | 100 | 100 | r.t. | 10 | 69% | 31% |
| 10 | 40 | 100 | 100 | 50 °C | 5 | 65% | 35% |
| 11 | 200 | 100 | 100 | r.t. | 5 | 46% | 40% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tisseraud, M.; Goutal, S.; Bonasera, T.; Goislard, M.; Desjardins, D.; Le Grand, R.; Parry, C.M.; Tournier, N.; Kuhnast, B.; Caillé, F. Isotopic Radiolabeling of the Antiretroviral Drug [18F]Dolutegravir for Pharmacokinetic PET Imaging. Pharmaceuticals 2022, 15, 587. https://doi.org/10.3390/ph15050587
Tisseraud M, Goutal S, Bonasera T, Goislard M, Desjardins D, Le Grand R, Parry CM, Tournier N, Kuhnast B, Caillé F. Isotopic Radiolabeling of the Antiretroviral Drug [18F]Dolutegravir for Pharmacokinetic PET Imaging. Pharmaceuticals. 2022; 15(5):587. https://doi.org/10.3390/ph15050587
Chicago/Turabian StyleTisseraud, Marion, Sébastien Goutal, Thomas Bonasera, Maud Goislard, Delphine Desjardins, Roger Le Grand, Chris M. Parry, Nicolas Tournier, Bertrand Kuhnast, and Fabien Caillé. 2022. "Isotopic Radiolabeling of the Antiretroviral Drug [18F]Dolutegravir for Pharmacokinetic PET Imaging" Pharmaceuticals 15, no. 5: 587. https://doi.org/10.3390/ph15050587